Research

MRSA accounts for over 100,000 bloodstream infections annually worldwide and over 20,000 of these infections result in death. Clinicians rely solely on intravenous courses of the antibiotic vancomycin to clear these infections. With such widespread use and reliance, the chances of vancomycin resistance occurring in MRSA is likely an inevitability with catastrophic effects.
I first began working in 2021 as a research scientist at University of Pittsburgh to understand if multi-drug resistant MRSA (methicillin-resistant Staphylococcus aureus) could develop the necessary genetic mutations to defeat the only antibiotic effective against MRSA, vancomycin, making it potentially invincible. We’re currently in the process of successfully evolving vancomycin resistance in MRSA. Then, through careful study in the lab, we can use these evolved superbugs to quantify the level of threat that vancomycin resistance poses to vulnerable patients in the clinic.
My secondary project looks at potentiation, or the underlying harmless mutations that in the right context can drive important factors like antibiotic resistance. I study potentiation in relation to the drug-bug combination of Zerbaxa and Psuedomonas aeruginosa. Through machine learning modeling of in vitro evolution, we seek to predict those infections where resistance will arise. Partnering with UPMC, our research is the future of applying personalized medicine approaches to patients in the infectious disease space.
In 2019 and 2020, I worked at the Rosenzweig Lab under Dr. Matthew Herron, studying the evolution of multicellularity in Chlamydomonas reinhardtii, a strain of alga. We observed its life cycle over the course of 6 days to understand cluster reproduction and phenotype.
In 2019, I studied the infection of avian influenza and seasonal influenza in lung tissue. My work identifying the colocalization of infection, Caspase-1 activation, and cell death in avian influenza-infected cells suggests pathways for the therapeautic treatment of influenza.
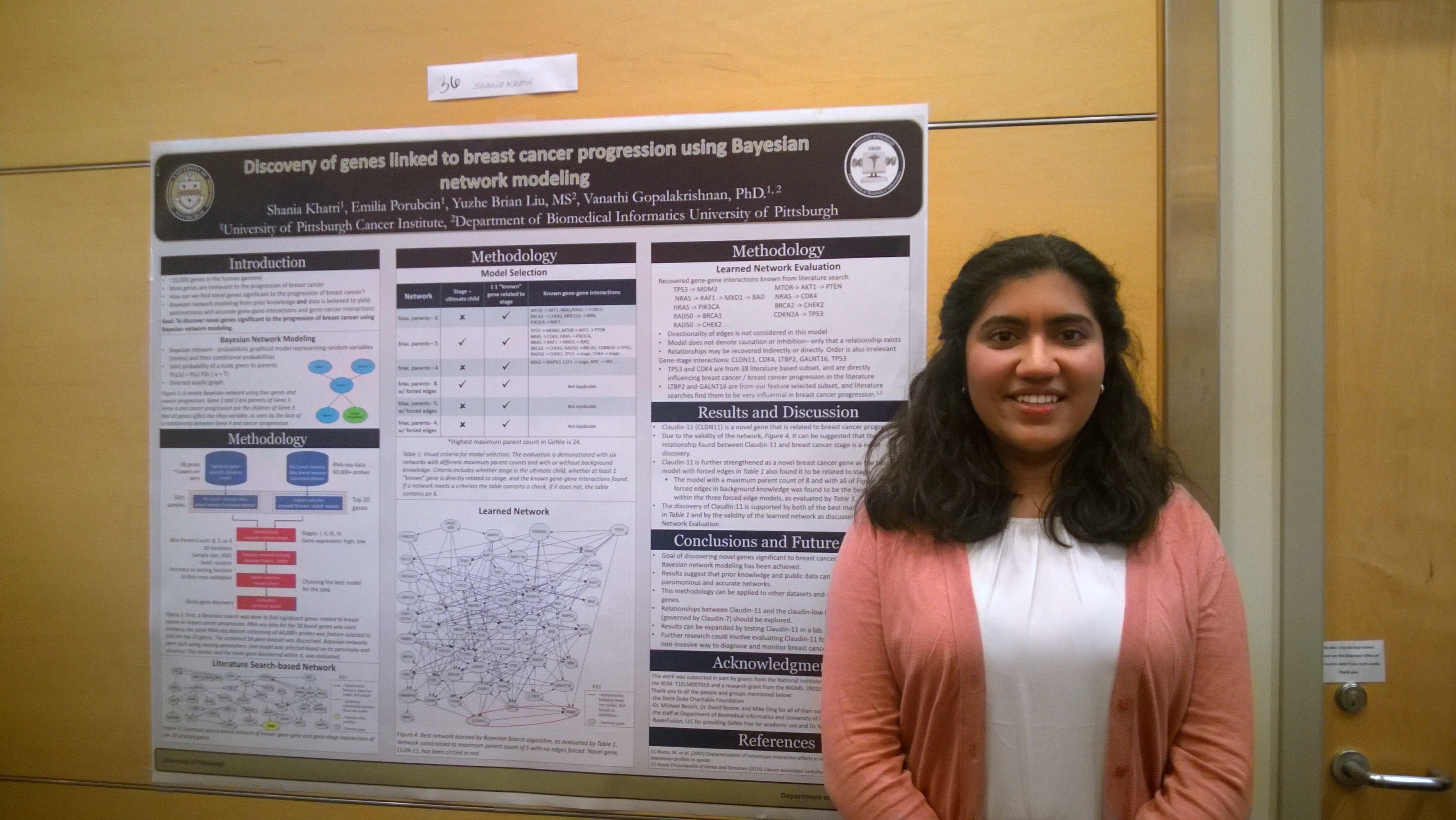
In 2016, I created a Bayesian framework to discover novel genes related to breast cancer progression. With this novel Bayesian framework, I was able to suggest gene CLDN11 as a human biomarker for breast cancer progression. I’ve been incredibly fortunate to have been able to present this work at the American Medical Informatics Association (AMIA)’s Annual 2016 Symposium in Chicago and Intel International Science and Engineering Fair (ISEF) 2017. You can read a short abstract of my presentation here.
In 2017, I built a pipeline for modeling community-acquired pneumonia. My model allows physicians to understand the clinical variables driving each patient’s illness and make informed decisions about the type of care given. I was given the opportunity to present this research at the AMIA Annual 2017 Symposium in Washington D.C.